Providing support to people affected by Turner Syndrome and their families in New Zealand
Providing support to people affected by Turner Syndrome and their families in New Zealand
Turner Syndrome is a medical condition named after the American physician, Dr. Turner, who in 1938 first fully described the condition.
Turner Syndrome affects only women. It is caused by a total or partial absence of one of the X chromosomes in some or all body cells. Turners Syndrome affects around 1 in 2500 live female births.
Syndrome means a collection of physical features which, when they occur together, help a doctor to recognise a particular condition. The most common physical features of Turner Syndrome include short stature and lack of sexual development at puberty. Other physical features that may occur include a low hair line, webbed neck and pigmented moles.
The diagnosis of Turner Syndrome can be confirmed through chromosome analysis, also called karyotype testing. This can be done either prior to birth through amniocentesis, or after birth at any stage through a simple blood test.
These things may sound daunting, but no one has all of these problems; most women have only a few, and all of them are able to be “managed”.
A multidisciplinary approach to treatment is important to improve the quality of life of girls and women with Turner Syndrome.
A congenital heart defect (CHD) occurs in approximately 30% of patients with Turner Syndrome. At the time of diagnosis, all individuals, regardless of age, should have a cardiac evaluation, including a complete physical examination and an echocardiogram. A cardiologist skilled in the assessment of congenital heart disease should interpret the echocardiogram. Blood pressure should be monitored at least annually in all patients with Turner Syndrome.
If a heart defect is present, the patient should be followed by a cardiologist in collaboration with the primary physician. If the initial cardiac evaluation from childhood does not show CHD, a repeat cardiovascular physical examination and echocardiogram, with particular attention paid to the aortic root, should be conducted at some time during adolescence (12–15 years of age).
Evaluation of renal anatomy. Congenital malformations of the urinary system are present in up to 30% of patients with Turner Syndrome most commonly horseshoe kidneys, a double collecting system, or abnormal vascular supply. Although many of these abnormalities do not have clinical significance, some may result in an increased risk of hypertension, urinary tract infections, or hydronephrosis. Therefore, all individuals with Turner Syndrome should have a renal ultrasound study performed at the time of diagnosis. If abnormalities are detected, further evaluations should be performed, and the appropriate therapy instituted. Additionally, in such individuals, ultrasound and urine cultures should be performed every 3–5 years .
Conductive and sensorineural hearing loss are common in girls with Turner Syndrome. The majority (50–90%) of women with Turner Syndrome have sensorineural hearing loss, manifest by a sensorineural dip in the 1.5–2 kHz region, sensorineural high frequency loss, or all of these. The sensorineural dip can occur as early as 6 years of age and occasionally leads to hearing impairment during childhood. The condition is progressive, however, and commonly leads to hearing problems in later life, which may have serious social consequences.
Otitis media (ear infection) is extremely common in girls with Turner Syndrome. It occurs particularly between 1 and 6 years of age, with a maximum incidence (>60%) at 3 years of age. The cause is still unknown, but growth retardation of the temporal bone may be important. Aggressive treatment of otitis media is appropriate, and insertion of ventilation tubes (grommets) should be considered. Careful follow-up is important. Patients with chronic middle ear problems should be operated on without delay to prevent repeat episodes. Short girls with extensive otitis media problems should be referred to an endocrinologist if Turner Syndrome has not previously been diagnosed.
Between 10–30% of individuals with Turner Syndrome develop primary hypothyroidism, generally associated with antithyroid antibodies. Often, there are no overt clinical symptoms. Levels of TSH and total or free T4 should therefore be measured at the time of diagnosis and at intervals of 1–2 years thereafter.
Girls with Turner Syndrome often have speech problems. If speech problems occur, referral to an ear, nose, and throat clinic and a speech therapist is recommended.
Strabismus (squint), amblyopia(failure of the retina to form sharp images), and ptosis(drooping of the eyelids) are common in Turner Syndrome. Ophthalmological evaluation should be part of the regular physical examination, with referral when appropriate.
Girls and women with Turner Syndrome have a predisposition to obesity, which may be exaggerated in appearance by the characteristic shield-like chest, stocky build, and short stature with relatively short legs. Individuals should be evaluated regularly, with appropriate counseling to avoid obesity.
Although there may be an increased risk of glucose intolerance, frank diabetes is rare in children with Turner Syndrome. Routine glucose tolerance tests are not necessary.
Infants with Turner Syndrome have an increased risk of congenital hip dislocation, which may be associated with degenerative arthritis of the hips in older women. Approximately 10% of girls with Turner Syndrome develop scoliosis, most commonly during adolescence. Evaluation for orthopedic problems should be part of the regular physical examination, with referral when appropriate.
The small lower jaw may contribute to overcrowded teeth and other dental abnormalities. An orthodontic examination should therefore be undertaken at 8–10 years of age.
Although most common in infants, lymphedema (swelling of hands and feet) may occur or reoccur at any age, and may be associated with the initiation of therapy with Growth Hormone or estrogen. Edema can usually be controlled with support stockings and/or diuretics.
The risk of keloid (overgrown thickened raised scar tissue) formation in Turner Syndrome is high. Elective surgery (e.g. for webbed neck or prominent ears) should be employed judiciously. This also applies to simple procedures, such as ear piercing.
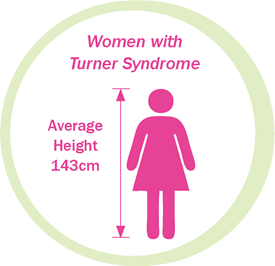
Surveys over the last 30 years have indicated that short stature affects at least 95% of all individuals with Turner Syndrome. Short stature is probably the most common, readily recognizable clinical feature of Turner Syndrome.
Growth hormone (GH) therapy is standard care for a child with Turner Syndrome and is usually begun after the child’s height falls below the fifth percentile for healthy girls in the same age group. More than one half of girls with Turner Syndrome will have fallen below the fifth percentile by 2 years of age. For those who experience early growth failure, it appears reasonable to begin GH therapy as early as 2 years of age.
GH increases the rate of growth in most girls with Turner Syndrome without advancing the bone age. Some reports indicate increases in final adult height of 8 to 10 cm if they receive at least 6 years of GH therapy and estrogen production is delayed. Greater height gains have recently been achieved even without delaying estrogen production by initiating GH at an early age and at high doses. Management of growth failure impacts on many other aspects of the care of individuals with Turner Syndrome, including estrogen replacement, socialization, and academic achievement. (Health Supervision)
Up to 30% of girls with Turner Syndrome will undergo spontaneous pubertal development, and 2–5% will have spontaneous periods and may have the potential to achieve pregnancy without medical intervention. Pubertal development may be delayed and, in most patients, is followed by progressive ovarian failure.
When estrogen therapy is required to induce pubertal development, the dosing and timing should be aimed at mimicking normal pubertal development, taking account of the individual’s desire to begin puberty and also of the family history of age at onset of puberty. Doses should be adjusted to the response of individual patients, which may be monitored in terms of the development of secondary sex characteristics, bone maturation, or uterine volume.
Estrogen therapy should be coordinated with the use of GH. This should be individualized for each patient so as to optimize both growth and pubertal development. When growth promotion is a priority, consideration should be given to delaying estrogen therapy to avoid compromising final height.
Estrogen therapy needs to be initiated and adjusted according to the needs and priorities of the individual with Turner Syndrome. Thus, if growth promotion is a priority, estrogen therapy should not be initiated before 12 years of age unless height has already been maximized. Estrogen therapy should ideally be started by 15 years of age.
Estrogen therapy should be initiated at a low dose (one sixth to one quarter of the adult dose) and increased gradually (at intervals of 3–6 months). Doses can then be adjusted to the response (Tanner stage, bone age, or uterine growth), with the aim of completing feminization gradually over a period of 2–3 years .
A progestin, such as medroxyprogesterone, should be added either when vaginal bleeding first occurs or after 12–24 months of estrogen therapy to establish monthly menstrual cycles.
Individuals with Turner Syndrome who have functioning ovaries and progress through puberty spontaneously should receive contraceptive and genetic counseling.
Adult women with Turner Syndrome should undergo a comprehensive medical evaluation due to the increased risk of a number of common diseases. All medical problems present during childhood should be followed in adults (e.g. CHD, hearing loss, skeletal problems, and dental and ophthalmological abnormalities). Annual medical history and general physical evaluation should be performed, including blood pressure, heart and blood pressure checks, clinical evaluation of thyroid size and function, breast examination, and Pap smear.
As in children, regular hearing examinations are important, as about 15% of adults with Turner Syndrome experience significant hearing loss, which may be conductive and/or sensorineural. The hearing loss is progressive, but tends to occur rapidly after about 35 years of age. Hearing aids are frequently necessary. If only a sensorineural dip is present, follow-up should occur every 3–5 years . Hearing assessments should be conducted every 10 years in patients who do not have hearing problems.
Many of the problems of adult life in patients with Turner Syndrome are compounded by obesity, partly due to low physical fitness. Lifestyle education with advice on diet and exercise must be included in a program of prevention of diabetes, osteoporosis, and hypertension. Women with Turner Syndrome should aim to have a body mass index below 25 kg/m2 and a waist/hip ratio less than 0.80.
Blood tests of women with Turner Syndrome should be carried out at 2-yr intervals and include measurements of hemoglobin, renal function (creatinine and blood urea nitrogen), fasting blood glucose, lipid profile, liver enzymes, and thyroid checks.
Individuals with known renal collecting system anomalies may require more frequent screening for urinary tract infections.
There is an increased incidence of all fractures in patients with Turner Syndrome over the age of 45 years . Measurements of bone mineral density should therefore be performed at the initial visit in adults with Turner Syndrome and 3–5 years later. If there is no change, further measurements can be made at less frequent intervals. If there is a significant deterioration in bone mass, current standards of practice for the treatment of osteoporosis should be instituted. An oral calcium intake of at least 1.2 g/day should be recommended, as should weight-bearing exercise.
Hypertension is more common in patients with Turner Syndrome than in the general population. Blood pressure should therefore be monitored routinely and hypertension treated vigorously with reference to age-specific normal ranges. Careful monitoring under the guidance of a cardiologist is advised before and during spontaneous or assisted pregnancy
Although a few patients with Turner Syndrome are able to achieve spontaneous pregnancy, most are infertile.
Various assisted reproductive techniques are now available for achieving pregnancy. Before contemplating pregnancy, either spontaneous or assisted, individuals with Turner Syndrome should undergo a complete medical evaluation.
Particular attention should be paid to the renal and cardiovascular systems, and thyroid status and glucose tolerance should be determined.
All pregnancies should be followed by a multidisciplinary team, including perinatologists, endocrinologists, and cardiologists.
Oocyte or embryo donation can be used to achieve pregnancy in patients with Turner Syndrome who do not have functional ovaries.
Special attention should be given to appropriate preparation of the uterus. This requires adequate hormone replacement therapy for 3–4 months before oocyte or embryo transfer, to increase the size of and improve the blood flow in the uterus.
Optimally, the thickness of the endometrium should be 7 mm. If this is not achieved using conventional hormone replacement therapy with daily estradiol and progesterone for 12 days in each cycle, the dose of estradiol should be increased.
Ideally, only one embryo should be transferred at a time, to avoid the additional risks associated with multiple pregnancies. Under optimal conditions, spontaneous vaginal delivery is an acceptable option. Cesarian section, however, is often employed because of a narrow pelvis.